Developing a Reproducible Workflow for Large-scale Phenotyping
Russell Poldrack
My name is Russell Poldrack, and I am a professor in the Department of Psychology at Stanford University. My work uses neuroimaging, genomics, and behavioral studies to examine the brain systems involved in decision making and executive control. Many of our workflows use high-performance computing due to the large data and complex nature of the workflows. This particular case study focuses on analysis of a study known as the "MyConnectome study", which involved intensive data collection from a single individual over the course of 18 months, including neuroimaging, genomic, metabolomic, and behavioral data. This large heterogenous dataset raised a number of new challenges for reproducible data analysis.
Workflow
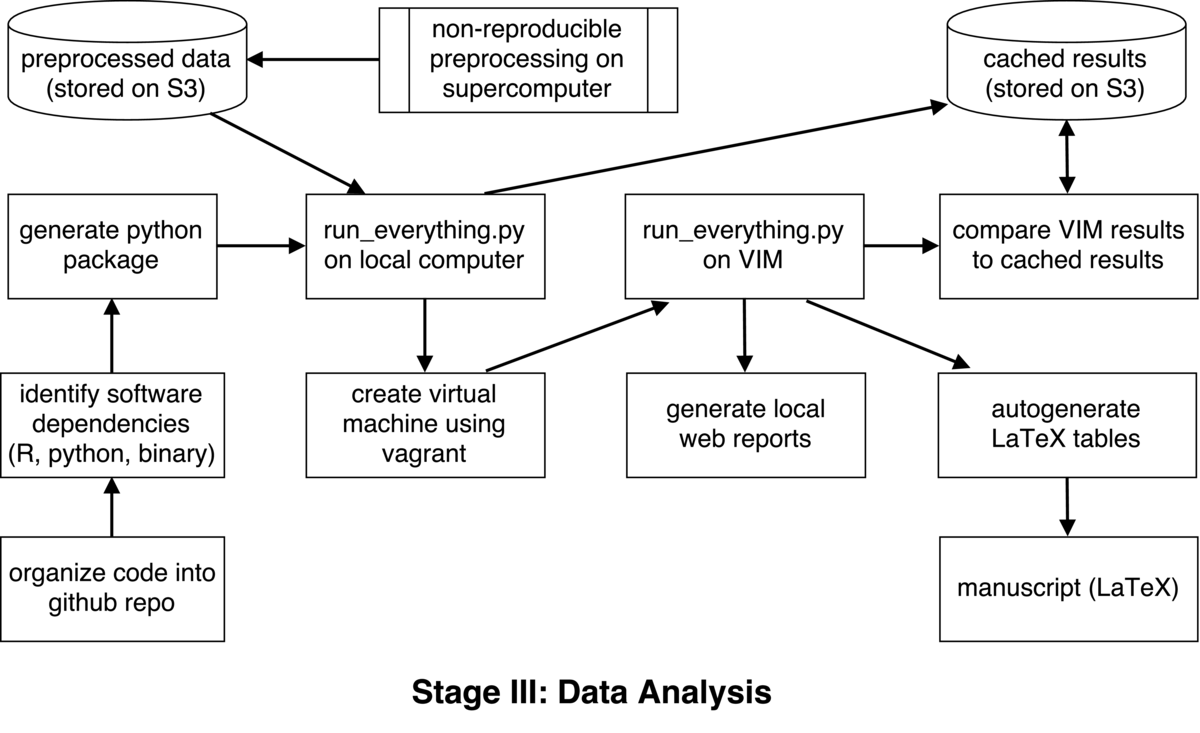 This workflow is meant to outline the analysis of a complex dataset including neuroimaging, behavioral, transcriptomic, and metabolomic data (http://www.myconnectome.org). The data were collected over the course of 18 months from a single individual, and will be made fully available online upon publication of the manuscript via the OpenFMRI project. The data are released under a public domain dedication, meaning that anyone can do anything they wish with the data, with no restrictions on redistribution or requirements of attribution. I chose this approach because I feel that it will provide greatest degree of utility for the data.
This workflow is meant to outline the analysis of a complex dataset including neuroimaging, behavioral, transcriptomic, and metabolomic data (http://www.myconnectome.org). The data were collected over the course of 18 months from a single individual, and will be made fully available online upon publication of the manuscript via the OpenFMRI project. The data are released under a public domain dedication, meaning that anyone can do anything they wish with the data, with no restrictions on redistribution or requirements of attribution. I chose this approach because I feel that it will provide greatest degree of utility for the data.
The data processing stream was initially built in a non-reproducible manner on a single laptop. After completing this, I became interested in generating a reproducible version of the workflow so that other researchers can exactly reproduce the analyses. A challenge of reproducible analysis in this study is that the raw data are very large (several TB including the raw genomic and neuroimaging data). These data are being made available to any who wishes to use them, but it is not possible to easily provide reproducible workflows for these operations because they require large-scale supercomputing resources. For the processes that require supercomputing (such as genome alignment for RNA-sequencing data and surface-based parcellation for MRI data), we have shared most of the code used to complete these workflows. Another complication of full reproducibility is that some of the preprocessing operations were performed by another laboratory, using code that they are not currently willing to share openly. Thus, we made the decision to focus on building an open reproducible workflow that encompasses as much as possible of the processing stream, using preprocessed data downloaded from an online archive.
The goal of this process was to create a completed automated analysis stream that requires no manual intervention. The workflow uses a number of tools, including python, R, MATLAB, and the Connectome Workbench (a domain-specific software tool for analysis of neuroimaging data). I started by generating scripts to perform each of the operations, but ultimately decided to generate a single python package to coordinate the entire workflow (available at https://github.com/poldrack/myconnectome). The first operation of this package is to download the preprocessed data from Amazon S3, using the boto package in python. We then perform additional processing of each of the different data types.
For the neuroimaging data, we developed a set of python functions to extract and summarize connectivity measures between different brain regions. These analyses included assessment of connectivity between regions using both standard correlation measures as well as regularized partial correlation (using the R QUIC package). Network analyses were performed using the Brain Connectivity Toolbox, and visualized using the Cytoscape software package.
For the transcriptome and metabolomic data, analyses were performed using a set of Rmarkdown scripts executed using R. These analyses included the identification of coexpression networks using the Weighted Gene Coexpression Network Analysis (WGCNA) package; eigengenes identified from these network were then used in subsequent analyses. The gene networks were annotated using DAVID. Metabolomic measures were clustered using affinity propagation, and annotated using IMPALA.
The outcomes of each of the foregoing analyses were saved and used to compute time series correlations between each of the measures across all domains (for a total of more than 20,000 statistical tests), using the R forecast package. These are then summarized in a web report generated using Rmarkdown. Currently the only test is one that compares the results of the full workflow to a set of results cached on S3. Documentation of the code is minimal.
After implementing the workflow on a single system, I then implemented it on a virtual machine in order to allow anyone anywhere to run it. I used the Vagrant software package to provision a virtual machine with all necessary requirements. Once installed, the user can run the entire workflow with a single command. In addition to running the entire data analysis workflow, the virtual machine also includes a web server that provides access to the results of all of the analyses, along with a data browser for the detailed results. This system is identical to the one exposed publicly at http://results.myconnectome.org. Documentation for installing and running the software is evolving.
Pain points
A number of pain points were encountered in the development of a reproducible analysis workflow. First, there were a number of processing stream operations that could not be implemented in this manner. In particular, some of the preprocessing operations required high-performance computing resources, which could not be generalized due to specifics of job submission systems. Second, there was a substantial amount of extra work necessary to generalize the code to work on an arbitrary system, primarily involving the identification and resolution of software dependencies. A third pain point involved the identification of appropriate technologies for sharing of a reproducible workflow. We have used a VM provisioned using Vagrant, but there are many other approaches that one might use. Finally, we struggled with identifying what level of user at whom we were targeting the project. We ultimately decided to make it easy to use for non-power-users, which required substantial extra work.
Key benefits
The reproducible workflow provides a number of important benefits. First, it provides a degree of detail that could not be feasibly included in the publication. Second, it increases the degree of trust amongst others in the field in the results that are presented in the publication. Third, it provides an example for others in our subfield of how to implement reproducible shared workflows.
Key tools
I have used Vagrant to allow any user to easily provision a virtual machine that includes all of the necesssary dependencies to run the workflow (see https://github.com/poldrack/myconnectome-vm).
Questions
What does "reproducibility" mean to you?
In the context of my case study, "reproducibility" means the ability to exactly reproduce the analysis workflow that was used to obtain the results reported in a manuscript. More generally, I take the term to also encompass the consistency of results across different workflows or datasets.
Why do you think that reproducibility in your domain is important?
The workflows used for neuroimaging data are highly complex with a great degree of analytic flexibility, which raises concerns regarding the reproducibility of results. We desperately need a greater degree of transparency in order to make research in our domain more reproducible.
How or where did you learn about reproducibility?
I think I primarily learned from negative examples; that is, from seeing other researchers whose research findings rely upon code that is not openly available and data that are not shared.
What do you see as the major challenges to doing reproducible research in your domain, and do you have any suggestions?
Human subjects issues and concerns about scooping are often raised here, but I think these are red herrings. The primary pitfall is that reproducible research practices make it harder to obtain splashy findings that get high-profile publications.
What do you view as the major incentives for doing reproducible research?
Increasing trust in one's research.
Are there any best practices that you'd recommend for researchers in your field?
Using version control software, and open sharing of data.